
The source of a protein is generally tissue or microbial cells. The first step in any protein purification procedure is to break open these cells, releasing their proteins into a solution called a crude extract. If necessary, differential centrifugation can be used to prepare sub-cellular fractions or to isolate specific organelles.
Commonly, the extract is subjected to treatments that separate the proteins into different fractions based on a property such as size or charge, a process referred to as fractionation. Early fractionation steps in a purification utilize differences in protein solubility, which is a complex function of pH, temperature, salt concentration, and other factors.
The solubility of proteins is lowered in the presence of some salts, an effect called ‘salting out’. The addition of certain salts in the right amount can selectively precipitate some proteins, while others remain in solution. Ammonium sulfate ((NH4)2SO4) is particularly effective and is often used to salt out proteins. The proteins thus precipitated are removed from those remaining in solution by low-speed centrifugation.
Dialysis is a procedure that separates proteins from small solutes by taking advantage of the proteins’ larger size. The partially purified extract is placed in a bag or tube made of a semipermeable membrane. When this is suspended in a much larger volume of buffered solution of appropriate ionic strength, the membrane allows the exchange of salt and buffer but not proteins. Thus dialysis retains large proteins within the membranous bag or tube while allowing the concentration of other solutes in the protein preparation to change until they come into equilibrium with the solution outside the membrane. Dialysis might be used, for example, to remove ammonium sulfate from the protein preparation.
Column Chromatography: A porous solid material with appropriate chemical properties (the stationary phase) is held in a column, and a buffered solution (the mobile phase) migrates through it. The protein, dissolved in the same buffered solution that was used to establish the mobile phase, is layered on the top of the column and allowed to percolate into the solid matrix by the addition of the buffer solution on top. The protein solution forms a band within the mobile phase that is initially the depth of the protein solution applied to the column. As proteins migrate through the column, they are retarded to different degrees by their different interactions with the matrix material. Individual types of proteins gradually separate from each other, forming bands within the broader protein band.
Separation improves (i.e., resolution increases) as the length of the column increases. However, each individual protein band also broadens with time due to diffusional spreading, a process that decreases resolution.
- Three types of column chromatographic methods used in protein purification.
- (a) Ion-exchange chromatography exploits differences in the sign and magnitude of the net electric charges of proteins at a given pH.
- (b) Size-exclusion chromatography, also called gel filtration, separates proteins according to size.
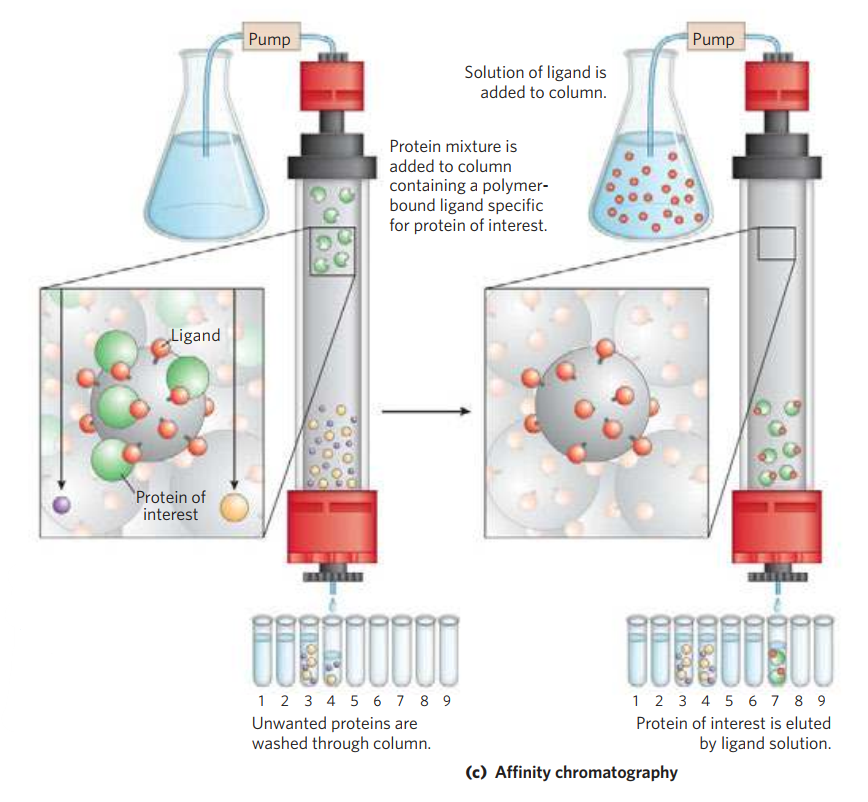
- (c) Affinity chromatography separates proteins by their binding specificities.
Ion-Exchange Chromatography: The column matrix is a synthetic polymer (resin) containing bound charged groups; those with bound anionic groups are called cation exchangers, and those with bound cationic groups are called anion exchangers. The affinity of each protein for the charged groups on the column is affected by the pH (which determines the ionization state of the molecule) and the concentration of competing free salt ions in the surrounding solution. Separation can be optimized by gradually changing the pH and/or salt concentration of the mobile phase so as to create a pH or salt gradient. In cation-exchange chromatography, the solid matrix has negatively charged groups. In the mobile phase, proteins with a net positive charge migrate through the matrix more slowly than those with a net negative charge, because the migration of the former is retarded more by interaction with the stationary phase.

Size-Exclusion Chromatography: In this method, large proteins emerge from the column sooner than small ones. The solid phase consists of cross-linked polymer beads with engineered pores or cavities of a particular size. Large proteins cannot enter the cavities and so take a shorter (and more rapid) path through the column, around the beads. Small proteins enter the cavities and are slowed by their more labyrinthine path through the column. Size-exclusion chromatography can also be used to approximate the size of a protein being purified similar to gel electrophoresis.

Affinity Chromatography: The beads in the column have a covalently attached chemical group called a ligand—a group or molecule that binds to a macromolecule such as a protein. When a protein mixture is added to the column, any protein with affinity for this ligand binds to the beads, and its migration through the matrix is retarded. For example, if the biological function of a protein involves binding to ATP, then attaching a molecule that resembles ATP to the beads in the column creates an affinity matrix that can help purify the protein. As the protein solution moves through the column, ATP-binding proteins (including the protein of interest) bind to the matrix. After proteins that do not bind are washed through the column, the bound protein is eluted by a solution containing either a high concentration of salt or free ligand—in this case, ATP or an analog of ATP. Salt weakens the binding of the protein to the immobilized ligand, interfering with ionic interactions. Free ligand competes with the ligand attached to the beads, releasing the protein from the matrix; the protein product that elutes from the column is often bound to the ligand used to elute it.
Chromatographic methods are typically enhanced by the use of HPLC, or high-performance liquid chromatography. HPLC makes use of high-pressure pumps that speed the movement of the protein molecules down the column, as well as higher-quality chromatographic materials that can withstand the crushing force of the pressurized flow. By reducing the transit time on the column, HPLC can limit diffusional spreading of protein bands and thus greatly improve resolution.

